GlycoEnzDB
Comprehensive glycoEnzyme database and dynamic pathway generation
Introduction ▲
Description: The GlycoEnzDB is a manually curated database that describes the complement of enzymes facilitating glycosylation in human systems. While a majority of the knowledge can be translated to other mammalian species, this is not always possible since some epitopes like Galα1,3Gal and monosaccharides like Neu5Gc are not found in humans. In total, the database describes 380+ manually curated glycogenes that regulate carbohydrate biosynthesis in humans, and upto 28 various pathway maps that these enzymes are involved in. . These data come from two reference texts in the field [1, 2], and other resources.
GlycoEnzDB has links to other databases related to nucleic acids [3, 4, 14], proteins [5, 16, 17], diseases [6], pathway maps [7, 15] and enzymes [8, 18]. Besides cataloging these links, a major thrust is to develop connections between these different elements by integrating textbook knowledge with wet-lab experimental data. Currently, this is done for high-throughput ChiP-Seq experiments that describe the transcription factor interactome regulating glycan biosynthesis [10] and micro RNA that regulate gene expression [11]. Glycosylation pathways are context/cell specific as the expression of the glycoEnzymes may vary with cell and tissue, and also the function of transcription factors and other regulatory elements may vary. To address this need to perform analysis in a context specific manner, we provide glycoEnzyme transcript data both for primary tissue [5] and for common cancer cell lines [13].
The overarching goal of this resource is to quantitatively relate different biological elements regulating carbohydrate biosynthesis using a Systems Glycobiology framework. This is done in GlycoEnzDB by incorporating the Glycosylation Network Analysis Toolbox (GNAT) into a web framework. This enables the creation and simulation of SBML format networks that can be simulated in a cell and context-specific manner ([19], details below).
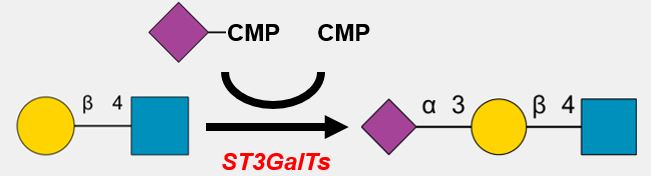
GlycoEnzDB facilities and links to literature: Search facilities enable search for enzymes based on 'Enzyme family' (pull down menu), gene symbol (e.g. 'ST3Gal4'), CAzy family (e.g 'GT29') and EC number (e.g. '2.4.99.2'). Hyperlinks are included to link individual enzyme definitions to figures in the Essentials of Glycobiology textbook [1].
Availability: Data related to the individual enzymes can be downloaded in .XML format. The full database can be downloaded as an XX file here.
Contact: For assistance related to this database, please contact Sriram Neelamegham (neel@buffalo.edu), Yusen Zhou (yusenzho@buffalo.edu) or Ted Groth (tgroth@buffalo.edu).
Acknowledgement: This site is supported by various grants from the National Institutes of Health.
Technical Details ▲
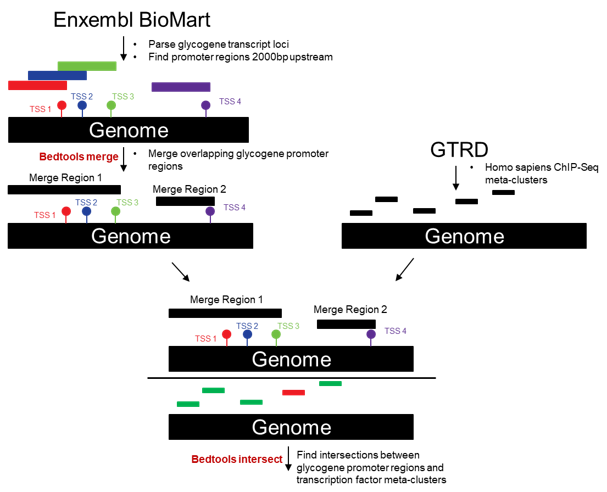
Transcription Factor ChiP-Seq data: The Gene Transcription Regulation Database (GTRD) were accessed to find transcription factor-to-glycogene relationships in ChIP-Seq data. The GTRD has analyzed publically available ChIP-Seq data with four separate algorithms: PICS, MACS, SISSRS, and GEM. The results from these four algorithms were agglomerated into a single result called a "meta-cluster". Meta-clusters are regions in the genome where multiple ChIP-Seq peak calling algorithms have predicted a transcription factor to bind. This meta-cluster information for Homo sapiens was downloaded from the GTRD as a tab-delimited file.
Transcription factors were deemed to potentially regulate glycogenes if the meta-cluster region overlapped with the promoter region of a glycogene. The promoter region of a glycogene was defined as 2000 bp upstream of the transcriptional start site (TSS). Glycogene coding region loci for every transcript variant of a glycogene were parsed from Ensembl's BioMart. Some promoter binding regions for glycogene transcript variants overlapped with one another. These regions were merged with one another for subsequent analysis using the bedtools "merge" function [12]. The glycogene promoter regions were intersected with GTRD's transcription factor meta-cluster regions using the bedtools "intersect" function. Only 1 base pair of overlap was required for intersection. The resulting table consisted of matched transcription factor meta-cluster locations and glycogenes
The GTRD also contains metadata concerning the biological system for which the transcription factor meta-clusters were identified, as well as any treatment conditions used in an experiment. In our database, we have parsed for experiments that have used cell lines from the Cancer Cell Line Encyclopedia (CCLE).
microRNA database: MicroRNA (miRNAs) are short noncoding (~22-nt) RNAs that repress translation or initiation mRNA degradation through binding to the 3’ UTR of mRNAs. Bound with an Argonaute protein to form a silencing complex, miRNAs function as sequence-specific guides, directing the silencing complex to transcripts, primarily through Watson-Crick pairing between the miRNA seed (miRNA nucleotides 2-7) and complementary sites within the 3’ untranslated regions (3’ UTRs) of the target RNAs. Analysis of preferentially conserved miRNA-pairing motifs has led to the identification of several classes of target sites including: i) 8mer site, which matches to miRNA positions 2-8 with an A opposite position 1, ii) 7mer-m8 site (position 2-8 match), iii) 7mer-A1 site (position 2-7 match with an A opposite position 1), iv) 6mer (position 2-7 match and v) offset-6mer (position 3-8 match) [9]. Recent work has shown miRNAs are major regulators of the glycome [20]. In several of these studies, inhibition of the glycogene recapitulates the biological effects of the miRNA.
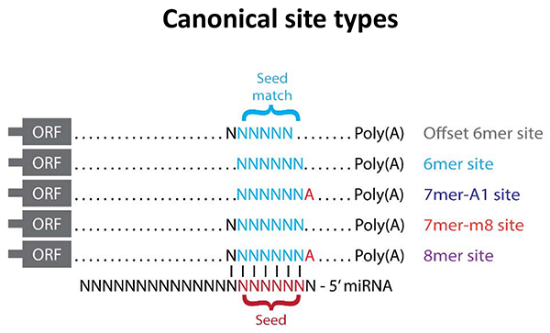
The predicted miRNA-glycoEnzyme interactome data are collected from the TargetScanHuman database [9]. TargetScan predicts biological targets of miRNAs by searching for the presence of 8mer, 7mer, and 6mer sites that match the seed region of each miRNA. In mammals, predictions are ranked based on the predicted efficacy of targeting as calculated using cumulative weighted context++ score of the sites. In TargetScanHuman7.0, a context++ model was developed combing unbiasedly selected 14 features to predict the targeting efficacy [9]. These 14 features include: site type, supplementary pairing, local AU, minimum distance, sRNA1A, sRNA1C, sRNA1G, sRNA8A, sRNA8C, sRNA8G, site8A, site8C, site8G, 3’ UTR length, SA, ORF length, ORF 8mer count, target site abundance, seed-pairing stability and probability of conserved targeting. In our database, the predicted conserved/non-conserved miRNA targets with context++ score ≤ -0.25 and context++ score percentile ≥ 90% are ranked and presented. The context++ score percentile rank is the percentage of sites for this miRNA with a less favorable context++ score. The sequence information in our database in based on the Genome Reference Consortium Human Build37 (GRCh37).
In miRNA-glycoEnzyme interactome section, a predicted targeting site map and corresponding table for list of the miRNAs are presented for selected glyco-enzyme. Click both miRNA name in figure or table, shown below, to further display detailed information including sequence alignment, site type, context++ score and etc.

GlycoEnzyme rules: The core feature of the GlycoEnzDB is the definition of the detailed enzyme specificity. Currently, all the 4 different types of glycosylation steps, N-/O-linked glycan, glycolipid, glycosaminoglycan and GPI-anchor, can be described using 3 fields of properties defined in the 'Enzyme Specificity' section. And all the values in the 'Enzyme Specificity' section use the standard IUPAC-condensed nomenclature with linkage information presented within parenthesis and branched structures enclosed in square brackets. These 3 fields are:
1. Substrate - This field describes the target monosaccharide that receives/loses child monosaccharide for the glycosyltransferase and glycosidase. And it also describes the target monosaccharide that is going to be replaced by the donor monosaccharide for epimerase.
2. Product - This field describes the substrate structure after receiving/losing or being replaced by the donor monosaccharide.
3. Constraints - Many of the glycoEnzymes are highly specific, which means they may have strict requirements for the substrates. This field is designed to meet these requirements. In Constraint field, it has 3 different types of sub-rules. They are as follows:
∙ Maximum/Minimum number of Sub-structure in substrate - which is used to describe the rules about number of certain substructures requiring for the substrate. For example, MAN1A1 can only act on glycans from M8 to M6. In this case, the number of alpha-mannose should between 5 and 7 for the substrate. This rule is defined as 'nMan(a1-?)≥5&nMan(a1-?)≤7', where the letter 'n' indicates that this constraint describes 'Maximum/Minimum number of Substructure' in substrate.
∙ Structure not allowed in substrate - which is used to describe the glycan structures that are not allowed in the substrate. There are two types of this 'not allowed structure' rules. First one is that this structure is not allowed in any position of the substrate, which is simply depicted by the symbol '!' followed by the 'not allowed structure'. For example, the B4GALT1 cannot add the Gal(b1-4) if the substrate structure contains Gal(b1-3), in this case which can be defined as '!Gal(b1-3)GlcNAc(b1-?)'. The second type focuses on the target branch in the substrate that receives/loses monosaccharide. In this case, a symbol '@' is used to describe the reaction site. For example, the B4GALT1 cannot add the Gal(b1-4) to the bisecting GlcNAc to form 'Gal(b1-4)GlcNAc(b1-4)…Man(b1-4) structure , which can be defined as '!@GlcNAc(b1-4)...Man(b1-4)'.
Instructions to create glycosylation pathway ▲
Select: Glycosylation pathways maps can be created for 4 types of carbohydrates: N-linked glycans, GalNAc type O-linked glycans, Glycosphingolipids and different types of glycosaminoglycans. This will select a subset of the relevant glycoEnzymes. Select your type. This narrow the list of Enzymes to be included in the pathway.
Demo: Clicking demo next to the glycan type, results in selection of the default enzyme list. This is a good point to start. Change the default enzyme selection now, using the checkboxes as necessary. This will tailor the enzymes involved in the formation of the core glycan structure ('initiation'), lactosamine chain 'extension', and chain 'termination'.
Compartment: Select the compartment(s) of the model, i.e. are all reactions going to be in one compartment or will you be interested in a multi-compartment model? Specify the residence time in each compartment.
Bracket: The output from pathway generation can be verbose with a number of isomeric structures. This presentation can be reduced by grouping isomeric glycans using brackets. Unselect the brackets if necessary.
Initial and final glycans: Specify either the 'initial glycan(s)' in the system or the 'final glycan(s)' or both. This covers three scenarios: 1. 'initial glycan only': Here, generates a generic network based on the selected glycoEnzymes starting with the ‘initial glycan’. Here, the 'production rate' of the initial glycan can also be specified. 2. 'final glycan only': The user has knowledge of the 'final glycan(s)' formed from say mass spectrometry analysis, and this is specified. Here, the initial glycan is the earliest glycan formed in the pathway. 3. 'initial and final glycan': In this scenario, the user has knowledge of the 'final glycan' and also wishes to constraint the 'initial glycan' and its 'production rate'. This will specific both the starting material and the final product in the network.
Network size: The network size can be limited by 'termination constraints' by: a. limiting the mass of the largest glycan formed in silico.; b. Specifying the maximum number of sub-structures formed in IUPAC format, e.g. to limit the number of sialic acids to 3 in the glycan, Sub-structure='Neu5Ac(a2-?)' and Max number =3; to limit Type II lactosamine chain number in glycan to 4, Sub-structure='Gal(b1-4)GlcNAc(b1-?)' and Max number=4; to also prevent longer lactosamine chains on a single antennae, to the previous case add a second constraint: Sub=structure='Gal(b1-4)GlcNAc(b1-3)Gal(b1-4)GlcNAc(b1-?)' and Max number=0.
Custom database generation ▲
The main GlycoEnzDB is dedicated to human glycosylation enzymes. If you are interested in creating a custom database for a different system, please write to Sriram Neelamegham (neel@buffalo.edu) for account access. We will also send you a custom database viewer and method to generate custom pathway maps.
Reference ▲
1. N. Taniguchi, et al. (2014) Handbook of Glycosyltransferases and Related Genes, Springer, Tokyo.
(link)
2. UniProt, C. (2019). "UniProt: a worldwide hub of protein knowledge." Nucleic acids research 47(D1): D506-D515.
(link)
3. Kanehisa, M. and S. Goto (2000). "KEGG: kyoto encyclopedia of genes and genomes." Nucleic acids research 28(1): 27-30.
(link)
4. Amberger, J. S., et al. (2015). "OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders." Nucleic acids research 43(Database issue): D789-D798.
(link)
5. Maglott, D., et al. (2011). "Entrez Gene: gene-centered information at NCBI." Nucleic acids research 39(Database issue): D52-D57.
(link)
6. Hansen, L., et al. (2015). "A glycogene mutation map for discovery of diseases of glycosylation." Glycobiology 25(2): 211-224.
(link)
7. Schomburg, I., et al. (2017). "The BRENDA enzyme information system-From a database to an expert system." Journal of Biotechnology 261: 194-206.
(link)
8. A. Varki, et al. (2017) Essentials of Glycobiology, Cold Spring Harbor (NY).
(link)
9. Agarwal, V., et al. "Predicting effective microRNA target sites in mammalian mRNAs. LID - 10.7554/eLife.05005 [doi] LID - e05005." (2050-084X (Electronic)).
(link)
10. Yevshin, I., et al. (2018). "GTRD: a database on gene transcription regulation—2019 update." Nucleic acids research 47(D1): D100-D105.
(link)
11. Yates, A. D., et al. (2019). "Ensembl 2020." Nucleic acids research 48(D1): D682-D688.
(link)
12. Quinlan, A. R. and I. M. Hall (2010). "BEDTools: a flexible suite of utilities for comparing genomic features." Bioinformatics (Oxford, England) 26(6): 841-842.
(link)
13. Barretina, J., et al. (2012). "The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity." Nature 483(7391): 603-607.
(link)
14. Pruitt, K. D., et al. (2007). "NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins." Nucleic acids research 35(Database issue): D61-D65.
(link)
15. Jassal, B., et al. (2019). "The reactome pathway knowledgebase." Nucleic acids research 48(D1): D498-D503.
(link)
16. Cantarel, B. L., et al. (2008). "The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics." Nucleic acids research 37(suppl_1): D233-D238.
(link)
17. Stelzer, G., et al. (2016). "The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses." Current Protocols in Bioinformatics 54(1): 1.30.31-31.30.33.
(link)
18. McDonald, A. G., et al. (2007). "ExplorEnz: a MySQL database of the IUBMB enzyme nomenclature." BMC Biochemistry 8(1): 14.
(link)
19. Liu, G., et al. (2012). "Glycosylation Network Analysis Toolbox: a MATLAB-based environment for systems glycobiology." Bioinformatics (Oxford, England) 29(3): 404-406.
(link)
20. Kurcon, T., et al. (2015). "miRNA proxy approach reveals hidden functions of glycosylation." Proceedings of the National Academy of Sciences 112(23): 7327-7332.
(link)